Molecular dynamics simulation can be thought of as a “computational microscope”, which is used to visualize the dynamic motion of molecules such as proteins and nuclear acids in our cells in new and in-depth ways. Their interactions regulate normal biological functions, while their dysfunction causes disease. Therefore, understanding these molecular mechanisms is important in both basic biology and applied medical research.
Modeling the dynamics of atoms
Molecular dynamics simulations have greatly deepened our understanding of important biological processes including protein folding, protein-drug binding, and protein-protein/nucleic acid interactions, which are often difficult to probe in experiments. Here, atoms and molecules are simulated to describe their motions and dynamics over time by resolving classical Newtonian physics so that motions of atoms in the molecules can be observed like in a collection of “movie frames”. To obtain accurate predictions, this requires that each step (time-step) must be very small, normally around femtosecond (10-15 s). However, important biological processes usually take place over milliseconds (10-3 s) and even longer time scales.
Even with world-class supercomputers, current molecular dynamics simulations can often reach only the microsecond (10-6 s) to millisecond (10-3 s) timescales. Simulating slower and very relevant biochemical processes occurring over milliseconds is very challenging through conventional molecular dynamics, which may take like 30 years to compute. Indeed, while more powerful computers would be needed, it would still remain a “grand challenge”.
GaMD brings molecular simulations to a step further
To bridge this big gap, a novel simulation method has been developed to accelerate molecular dynamics simulations. This technique — named Gaussian accelerated molecular dynamic (GaMD) — can accelerate molecular dynamics simulations thousands to tens of millions of times. It works by smoothing the potential energy surface and reducing the energy barriers. GaMD has been established for advanced simulation studies of a wide range of biomolecular systems, from proteins to membranes, carbohydrates, enzymes and nucleic acids, which makes GaMD a “Genius” which can enable the description of biophysical processes that were previously difficult to model.
One prominent application concerns the CRISPR-Cas9 system, which is a leading genome editing tool.
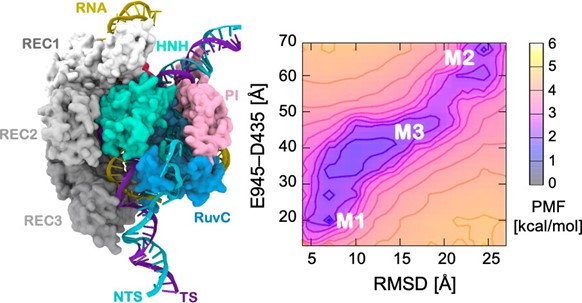
Assessing the dynamics and complex conformational transitions of CRISPR–Cas9 systems has been particularly challenging but understanding them is important to understand the mechanism of genome editing. For example, when editing the genome, the Cas9 protein is tightly bound to DNA and RNA and simulating the complex interplay of interactions between protein and nucleic acids during this process could not be done using conventional computational methods — it required the accelerating power of GaMD.
Microsecond GaMD simulations were able to describe the conformational transitions that enable the CRISPR protein to bind and process nucleic acids, with unprecedented dynamic details that scientists were previously unable to capture. Impressively, GaMD predicted the structure of the active complex before the experimental characterization, which coincided and confirmed the ability of GaMD to reliably capture structural transitions in biomolecules.
Not only that, GaMD was also used to investigate the onset of off-target effects in CRISPR-Cas9, a severe issue where the system recognizes and cleaves unwanted DNA sequences, resulting in errors in genome editing—with potentially hazardous consequences for the health or stability of the organism or system being edited. GaMD was used to describe how off-target DNA sequences bind the Cas9 protein, thereby providing information of the fundamental mechanisms behind these effects, allowing scientists to modify and optimize future CRISPR technologies to avoid these detrimental errors.
Within the context of the current COVID-19 pandemic, computer modeling has the potential to identify key viral interactions that could help researchers to develop new medicines and vaccines faster than manual drug searchers. GaMD simulations in particular has been used to design drug candidates that could block the main protease of the SARS-CoV-2 virus, an important target in the fight against COVID-19. In addition, a recently developed algorithm called selective Ligand GaMD (LiGaMD) captured, for the first time, both the binding and dissociation of an inhibitor to the human angiotensin-converting enzyme 2 (ACE2), the receptor hijacked by SARS-CoV-2 for virus entry into host cells. These simulations have provided important insights into functional mechanisms of the proteins that would otherwise not be possible, which will facilitate rational drug design for therapeutic treatments of COVID-19 and other important diseases.
An important step forward, continued innovations in both supercomputing hardware and method developments may help us to address increasingly challenging problems in the future, providing alternative means to solve problems on a molecular level.
Reference: Jinan Wang, et al., Gaussian accelerated molecular dynamics: Principles and applications, WIREs Computational and Molecular Science (2021). DOI: 10.1002/wcms.1521