
Binary semiconductors made out of elements from III and VI columns of the periodic table, like InSe and GaSe, have a tendency to crystallize in the form of double layers, whereby the cations stick “back to back”, each one holding three anions outside, to make outer surfaces of the layer. Adjacent layers (of hexagonal structure) are weakly coupled and can be piled with a different offset, giving rise to various polytypes. Although the energies, and hence the expected stabilities, of different polytypes seem to be very close, one particular phase (gamma) seems to be pronouncedly dominant in InSe, whereas a different other one (beta) – in GaSe.
The tiny energy differences that bring about such structural preferences were studied, in the work by J. Srour and colleagues, within density functional theory calculations “from first principles” probing the variation of energy as the volume of unit cell was compressed or expanded, permitting also the relaxation of the internal parameters (bond lengths). The energy closeness between the polytypes imposed a need for the utmo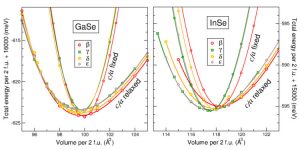 st numerical accuracy that could have been indeed provided by using (in comparison) two well proven computer codes, WIEN2k and VASP, broadly applied for electronic structure and total energy calculations. The “standard” modern versions of generalized gradient approximation for the exchange-correlation, a “working horse” of the practical density functional theory, have been used.
st numerical accuracy that could have been indeed provided by using (in comparison) two well proven computer codes, WIEN2k and VASP, broadly applied for electronic structure and total energy calculations. The “standard” modern versions of generalized gradient approximation for the exchange-correlation, a “working horse” of the practical density functional theory, have been used.
The study of J. Srour et al. extends, further on, onto the compounds of the same chemical composition as InSe and GaSe, but of different stoichiometry, namely, In2Se3 and Ga2Se3. For these systems, again, different structural arrangements seem plausible, and could have been verified and tested with the help of first-principles calculations. Specifically, the layered structures, in which the inner cation-cation bonds were cut by inserting an extra anion layer, were compared to bulk wurtzite-type structure with spatially distributed (and ordered) cation vacances, and found to be energetically higher than the latter. These findings throw some light onto the hitherto disputable structure models.
Text kindly provided by the authors J. Srour, A. Postnikov, M. Badawi, and F. El Haj Hassan. Image credit: Georgy Shafeev/Shutterstock








