Image credit: Ga on Unsplash
Natural products are chemical compounds that are produced by a living organism. They provide inspiration for new medicines and form the basis of over a third of all drugs approved by the United States’ Food and Drug Administration (FDA).
Countless natural products exist in nature, however, predicting how they might behave as a medicine or what they might do once inside the human body is difficult without intensive study. Many of those with medicinal properties are only found in tiny quantities in nature, which makes them hard to harvest in useful amounts, and they are usually complicated to create in a laboratory. This makes it challenging to find and test new natural products for medicine. A research team led by Professor Gisbert Schneider of ETH Zurich have designed a computational method to make this easier.
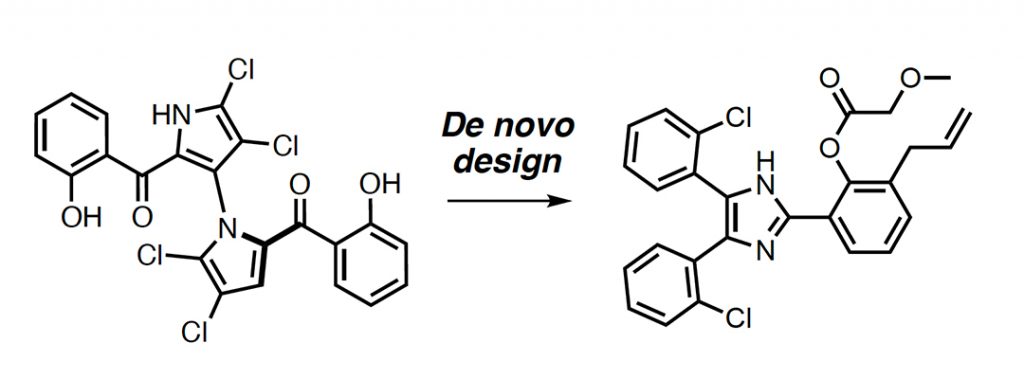
As proof-of-concept, Schneider and colleagues chose the known natural product, Marinopyrrole A, found in some marine bacteria and has established antibacterial and anticancer properties. To date, the shortest reported synthetic route for this molecule has five steps and yields 16% product based on the starting material used (not bad for a natural product synthesis, which sometimes require upwards of 20 steps and with even lower yields). Using their computational method, the team aimed to design de novo (literally meaning ‘of new’) compounds that are related to Marinopyrrole A, but that would be easier to synthesize.
The researchers used an algorithm called the “design of genuine structures”, or DOGS, to generate these new structures. The algorithm draws from a library of possible chemical transformations and uses this to build new compounds that are similar to the template compound — in this case Marinopyrrole A.
Because the DOGS algorithm generates molecules in a step-wise fashion, it can also suggest possible methods for synthesizing the compounds in a laboratory. A total of 802 de novo designs were generated for Marinopyrrole A, all of which required no more than three steps to create.
After the DOGS algorithm had been run, the next step was to analyze the new compounds using the chemically advanced template search (CATS) metric. The acronyms were coined by Schneider to avoid technical terms when communicating with non-specialists. “It all started with the CATS software more than two decades ago — I often programmed with my cat on the lap or the desk,” said Schneider in an email.
The CATS method ranked all 802 compounds based on their similarity to Marinopyrrole A. Compounds that are very similar are “conservative” and more likely to act in a similar way. Compounds that are more dissimilar are “explorative” because their effects are harder to predict.
To corroborate the algorithm’s findings, the team chose two of these 802 compounds to synthesize. First, to see if the synthetic route proposed by DOGS would work, and second, to test their biological activity.
By following the steps proposed by DOGS, they were able to synthesize the molecules and achieved yields of up to 66% — quite a feat in natural product synthesis, just ask any grad student.
The biological activity of the compounds was then predicted by SPiDER software, which compares the chemical structures with that of known drugs.
“The basic assumption is that molecules that have similar ‘pharmacophore’ patterns and properties also have similar bioactivity, that is, they bind to similar targets,” said Schneider. “SPiDER software uses a special kind of neural network and statistical methods to decide if the computed similarity between a query molecule and known drugs is significant. If so, the algorithm suggests some of the known targets of the drugs that are similar to the query molecule.”
When tested in the lab, the compounds showed similar activity to nonsteroidal anti-inflammatory drugs widely used in medicine, aligning with predictions made by SPiDER.
Given how incredibly difficult and expensive it is to find, create, and test natural product compounds normally, the DOGS and CATS methods could save research teams many years and hundreds of thousands of dollars when designing potential drug compounds.
Professor Schneider was not surprised by how well the DOGS and CATS computer programs worked. “There is ample evidence that these methods work reliably,” he said. “Not every design is a hit, though. The fact that the software generated a molecule with such exceptional bioactivity from scratch was a welcome surprise, given that the tools do not quantitatively predict activity.”
In the future, this could help researchers to find and create new medicines in a more environmentally and economically sustainable way, as well as more rapidly — which is particularly important for highly contagious diseases such as COVID-19.
“We are in the process of systematically generating synthetically accessible de novo designs for all known bioactive natural products,” said Schneider. “[In the future], we hope to contribute to obtaining ‘better drugs faster’ by learning from natural products with AI.”
Reference: L. Friedrich, et al., Learning from Nature: From a Marine Natural Product to Synthetic Cyclooxygenase-1 Inhibitors by Automated De Novo Design, Advanced Science (2021). DOI: doi.org/10.1002/advs.202100832