Caused by nonsense mutations in the dystrophin gene, Duchenne Muscular Dystrophy is a fatal, muscle-wasting disease affecting roughly one in 5,000 boys. These mutations prevent the body from producing functional, full-length proteins—in this case dystrophin proteins—by sending premature signals to stop production. While Duchenne can be diagnosed at birth, it is usually discovered between ages 2–5 and untreated patients often require wheelchairs by ages 10–12. The mean survival rate is only 19 years, as no cure exists.
Currently, glucocorticoids, a class of steroid hormones, are the most widely used medication for Duchenne and can increase a lifespan by up to three years—at a devastating cost. Its adverse effects include vertebral fracture, growth retardation, cataracts, Cushingoid features, and excessive weight gain. Researchers are now shifting focus from steroids to drug delivery systems, aiming to replace and repair the mutated dystrophin gene.
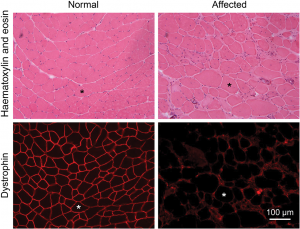
Skeletal muscle cross-sections from a normal and an affected mouse
In the review article “Nanotherapy for Duchenne muscular dystrophy” published in WIREs Nanomedicine and Nanobiotechnology, Michael Nance and colleagues from the University of Missouri’s Columbia School of Medicine discuss emerging nanomedications and therapies with potential to alter Duchenne’s fatal outcome. Restoring dystrophin expression in muscle tissue is key to these endeavors. This is complicated by the fact that the dystrophin gene is one of the largest in the genome, and the muscle the largest tissue in the body, while nanoparticles are inherently limited by their size.
Scientists previously doubted the possibility of a full body treatment, but several Adeno-associated virus (AAV) serotypes were tested for intravenous delivery in 2004. These viruses can insert genetic material in a specific location on a chromosome with near-certainty, although they can only carry a small amount of material. Many are still in early development, but some are now advancing for phase I clinical trials. These include Ataluren in Europe and Exondys51 in the USA.
Ataluren is a “read-through” drug, which works by “reading through” the premature stop signals put out by nonsense mutations and allowing the cell to continue producing a full-length, functional protein. Exondys51 is an “exon-skipping” drug, which causes cells to “skip” mutated areas of the genetic code. These trials will offer critical insights on moving Duchenne nanotherapy from mice and dogs to human patients. The review’s authors are hopeful these creative approaches will result in therapy breakthroughs.