It should not surprise us that even in parts of the genome where we don’t obviously see a “functional” code (i.e., one that’s been evolutionarily fixed as a result of some selective advantage), there is a type of code, but not like anything we’ve previously considered as such. And what if it were doing something in three dimensions as well as the one dimension of the ATGC code? A paper just published in BioEssays explores this tantalizing possibility…
Isn’t it wonderful to have a really perplexing problem to gnaw on, one that generates almost endless potential explanations. How about “what is all that non-coding DNA doing in genomes?”—that 98.5% of human genetic material that doesn’t produce proteins. To be fair, the deciphering of non-coding DNA is making great strides via the identification of sequences that are transcribed into RNAs that modulate gene expression, may be passed on transgenerationally (epigenetics) or set the gene expression program of a stem cell or specific tissue cell. Massive amounts of repeat sequences (remnants of ancient retroviruses) have been found in many genomes, and again, these don’t code for protein, but at least there are credible models for what they’re doing in evolutionary terms (ranging from genomic parasitism to symbiosis and even “exploitation” by the very host genome for producing the genetic diversity on which evolution works); incidentally, some non-coding DNA makes RNAs that silence these retroviral sequences, and retroviral ingression into genomes is believed to have been the selective pressure for the evolution of RNA interference (so-called RNAi); repetitive elements of various named types and tandem repeats abound; introns (many of which contain the aforementioned types of non-coding sequences) have transpired to be crucial in gene expression and regulation, most strikingly via alternative splicing of the coding segments that they separate.
Still, there’s plenty of problem to gnaw on because although we are increasingly understanding the nature and origin of much of the non-coding genome and are making major inroads into its “function” (defined here as evolutionarily selected, advantageous effect on the host organism), we’re far from explaining it all, and—more to the point—we’re looking at it with a very low-magnification lens, so to speak. One of the intriguing things about DNA sequences is that a single sequence can “encode” more than one piece of information depending on what is “reading” it and in which direction – viral genomes are classic examples in which genes read in one direction to produce a given protein overlap with one or more genes read in the opposite direction (i.e., from the complementary strand of DNA) to produce different proteins. It’s a bit like making simple messages with reverse-pair words (a so-called emordnilap). For example: REEDSTOPSFLOW, which, by an imaginary reading device, could be divided into REED STOPS FLOW. Read backwards, it would give WOLF SPOTS DEER.
Now, if it is of evolutionary advantage for two messages to be coded so economically – as is the case in viral genomes, which tend to evolve towards minimum complexity in terms of information content, hence reducing necessary resources for reproduction—then the messages themselves evolve with a high degree of constraint. What does this mean? Well, we could word our original example message as RUSH-STEM IMPEDES CURRENT, which would embody the same essential information as REED STOPS FLOW. However, that message, if read in reverse (or even in the same sense, but in different chunks) does not encode anything additional that is particularly meaningful. Probably the only way of conveying both pieces of information in the original messages simultaneously is the very wording REEDSTOPSFLOW: that’s a highly constrained system! Indeed, if we studied enough examples of reverse-pair phrases in English, we would see that they are, on the whole, made up of rather short words, and the sequences are missing certain units of language such as articles (the, a); if we looked more closely, we might even detect a greater representation than average of certain letters of the alphabet in such messages. We would see these as biases in word and letter usage that would, a priori, allow us to have a stab at identifying such “dual-function” pieces of information.
Now let’s return to the “letters”, “words”, and “information” encoded in genomes. For two distinct pieces of information to be encoded in the same piece of genetic sequence we would, similarly, expect the constraints to be manifest in biases of word and letter usage—the analogies, respectively, for amino acid sequences constituting proteins, and their three-letter code. Hence a sequence of DNA can code for a protein and, in addition, for something else. This “something else”, according to Giorgio Bernardi, is information that directs the packaging of the enormous length of DNA in a cell into the relatively tiny nucleus. Primarily it is the code that guides the binding of the DNA-packaging proteins known as histones. Bernardi refers to this as the “genomic code”—a structural code that defines the shape and compaction of DNA into the highly-condensed form known as “chromatin”.
But didn’t we start with an explanation for non-coding DNA, not protein-coding sequences? Yes, and in the long stretches of non-coding DNA we see information in excess of mere repeats, tandem repeats and remnants of ancient retroviruses: there is a type of code at the level of preference for the GC pair of chemical DNA bases compared with AT. As Bernardi reviews, synthesizing his and others’ groundbreaking work, in the core sequences of the eukaryotic genome, the GC content in structural organizational units of the genome termed “isochores” increased during the evolutionary transition between so-called cold-blooded and warm-blooded organisms. And, fascinatingly, this sequence bias overlaps with sequences that are much more constrained in function: these are the very protein-coding sequences mentioned earlier, and they—more than the intervening non-coding sequences—are the clue to the “genomic code”.
Protein-coding sequences are also packed and condensed in the nucleus – particularly when they’re not “in use” (i.e., being transcribed, and then translated into protein) – but they also contain relatively constant information on precise amino acid identities, otherwise they would fail to encode proteins correctly: evolution would act on such mutations in a highly negative manner, making them extremely unlikely to persist and be visible to us. But the amino acid code in DNA has a little “catch” that evolved in the most simple of unicellular organisms (bacteria and archaea) billions of years ago: the code is partly redundant. For example, the amino acid Threonine can be coded in eukaryotic DNA in no fewer than four ways: ACT, ACC, ACA or ACG. The third letter is variable and hence “available” for the coding of extra information. This is exactly what happens to produce the “genomic code”, in this case creating a bias for the ACC and ACG forms in warm-blooded organisms. Hence, the high constraint on this additional “code”—which is also seen in parts of the genome that are not under such constraint as protein-coding sequences—is imposed by the packaging of protein-coding sequences that embody two sets of information simultaneously. This is analogous to our example of the highly-constrained dual-information sequence REEDSTOPSFLOW.
Importantly, however, the constraint is not as strict as in our English language example because of the redundancy of the third position of the triplet code for amino acids: a better analogy would be SHE*ATE*STU* where the asterisk stands for a variable letter that doesn’t make any difference to the machine that reads the three-letter component of the four-letter message. One could then imagine a second level of information formed by adding “D” at these asterisk points, to make SHEDATEDSTUD (SHE DATED STUD). Next imagine a second reading machine that looks for meaningful phrases of a “sensitive nature” containing a greater than average concentration of Ds. This reading machine carries a folding machine with it that places a kind of peg at each D, kinking the message by 120 degrees in a plane. a point where the message should be bent by 120 degrees in the same plane, we would end up with a more compact, triangular, version. In eukaryotic genomes, the GC sequence bias proposed to be responsible for structural condensation extends into non-coding sequences, some of which have identified activities, though less constrained in sequence than protein-coding DNA. There it directs their condensation via histone-containing nucleosomes to form chromatin.
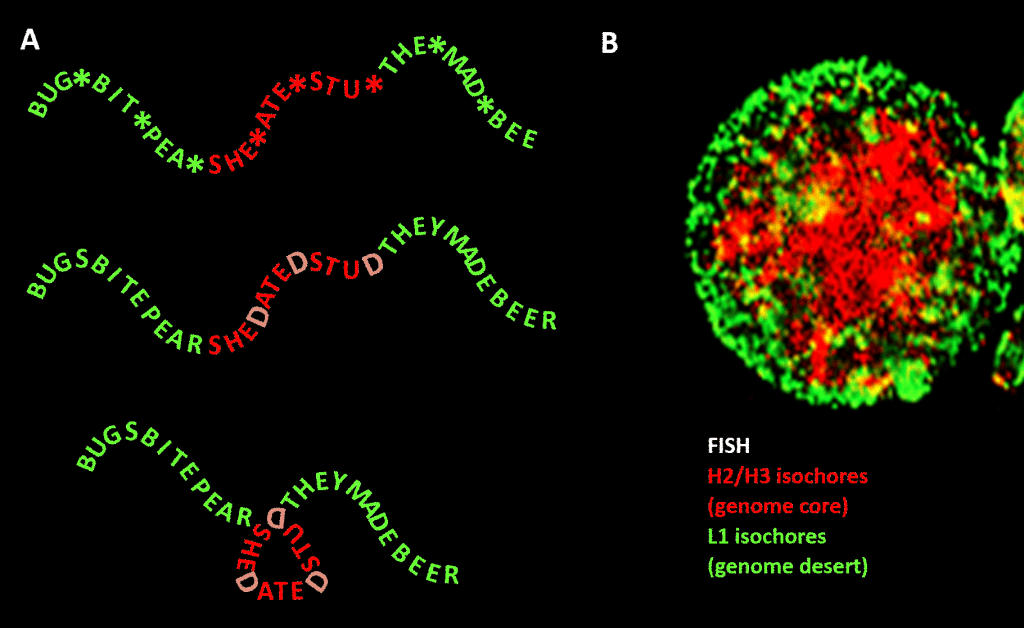
Figure. Analogy between condensation of a word-based message and condensation of genomic DNA in the cell nucleus. Panel A: Information within information, a sequence of words with a variable fourth space which, when filled with particular letters, generates a further message. One message is read by a three-letter reading machine; the other by a reading machine that can interpret information extending to the 4th—variable—position of the sequence. The second reader recognizes “sensitive” information that should be concealed, and at the points where a “D” appears in the 4th position, it folds the string of words, hence compressing the “sensitive” part and taking it out of view. This is an analogy for the principle of genomic 3D compression via chromatin, as depicted in panel B: a fluorescence image (via Fluorescence In-Situ Hybridization – FISH) of the cell nucleus. H2/H3 isochores, which increased in GC content during evolution from cold-blooded to warm-blooded vertebrates, are compressed into a chromatin core, leaving L1 isochores (with lower GC content) at the periphery in a less-condensed state. The “genomic code” embodied in the high-GC tracts of the genome is, according to Bernardi [1], read by the nucleosome-positioning machinery of the cell and interpreted as sequence to be highly compressed in euchromatin. Acknowledgements: Panel A: concept and figure production: Andrew Moore; Panel B: A FISH pattern of H2/H3 and L1 isochores from a lymphocyte induced by PHA—courtesy of S. Saccone—as reproduced in Ref. [1].]
These regions of DNA may then be regarded as structurally important elements in forming the correct shape and separation of condensed coding sequences in the genome, regardless of any other possible function that those non-coding sequences have: in essence, this would be an “explanation” for the persistence in genomes of sequences to which no “function” (in terms of evolutionarily-selected activity), can be ascribed (or, at least, no substantial function).
A final analogy—this time much more closely related—might be the very amino acid sequences in large proteins, which do a variety of twists, turns, folds etc. We may marvel at such complicated structures and ask “but do they need to be quite so complicated for their function?” Well, maybe they do in order to condense and position parts of the protein in the exact orientation and place that generates the three-dimensional structure that has been successfully selected by evolution. But with a knowledge that the “genomic code” overlaps protein coding sequences, we might even start to become suspicious that there is another selective pressure at work as well…
Andrew Moore, Ph.D.
Editor-in-Chief, BioEssays
Reference: G. Bernardi. 2019. The genomic code: a pervasive encoding/moulding of chromatin structures and a solution of the “non-coding DNA” mystery. BioEssays 41:12. 1900106